Подострая некротизирующая энцефаломиопатия Лея
О заболевании впервые было упомянуто в 1951 г. К настоящему времени описано более 120 случаев. Болезнь Лея (OMIM 256000) - генетически гетерогенное заболевание, которое может наследоваться как по ядерному типу (аутосомно-рецессивно или сцепленно с Х-хромосомой), так и митохондриально (реже).
Код по МКБ-10
Причины синдрома Лея
В основе заболевания лежит дефицит ферментов, обеспечивающих образование энергии главным образом за счёт нарушения обмена пировиноградной кислоты и дефекта транспорта электронов в дыхательной цепи. Развивается дефицит пируватдегидрогеназного комплекса (а-Е1-субъединицы), пируваткарбоксилазы, комплекса 1 (НАД-коэнзим Q-редуктаза) и комплекса 4 (цитохромоксидаза) дыхательной цепи.
При этом установлено, что дефекты пируваткарбоксилазы, комплекса 1 (НАД-коэнзим Q-редуктаза) и комплекса 4 (цитохромоксидаза) дыхательной цепи наследуются по аутосомно-рецессивному типу, дефекты пируватдегидрогеназного комплекса (а-Е1-субъединицы) - Х-сцепленно рецессивно. При точковых мутациях мтДНК, которые затрагивают 6-ю субъединицу АТФазы, характерно митохондриальное наследование. Чаще всего происходит мисценс-мутация, связанная с заменой тимина на гуанин или цитозин в положении 8993 мтДНК. Реже встречается мутация в позиции 9176 мтДНК. В связи с тем что мутация T8993G - основной дефект при синдроме NARP, описаны семьи с наличием этих двух заболеваний. У детей описана также мутация мтДНК в позиции 8344, которая встречается при синдроме MERRF.
Предполагают, что в случае накопления мутантной мтДНК в большинстве митохондрий развивается тяжёлое течение синдрома Лея. При митохондриальном генезе этого состояния мутантную мтДНК обнаруживают в 90% всех митохондрий. Патогенез связан с нарушением образования энергии в клетках и развитием лактат-ацидоза.
Симптомы синдрома Лея
Первые признаки заболевания дебютируют в раннем возрасте (1-3 года). Однако известны случаи манифестации болезни в 2-недельном и в 6-7-летнем возрасте. Вначале развиваются неспецифические нарушения: задержка психомоторного развития, снижение аппетита, эпизоды рвоты, дефицит массы тела. В последующем нарастают неврологические симптомы: мышечная гипотония или дистония с переходом в гипертонус, приступы миоклонии или тонико-клонические судороги, тремор конечностей, хореоатетоз, расстройство координации, снижение сухожильных рефлексов, вялость, сонливость. Церебральная нейродегенерация носит прогрессирующий характер. Нарастают симптомы пирамидной и экстрапирамидной недостаточности, нарушается акт глотания. Нередко наблюдают такие изменения органа зрения, как птоз, офтальмоплегия, атрофия зрительных нервов, реже пигментная дегенерация сетчатки. Иногда развиваются гипертрофическая кардиомиопатия, появляются эпизоды тахипноэ.
Редко заболевание протекает по типу острой энцефалопатии. Более характерно хроническое или подострое течение, которое приводит к летальному исходу через несколько лет после начала заболевания. При быстром течении (несколько недель) смерть наступает в результате паралича дыхательного центра.
Диагностика синдрома Лея
При биохимическом исследовании крови выявляют лактат-ацидоз вследствие накопления молочной и пировиноградной кислот в крови и ликворе, а также увеличение содержания аланина в крови. Также может быть повышен уровень кетоновых тел. В моче выявляют повышенную экскрецию органических кислот: молочной, фумаровой и др. Часто снижается уровень карнитина в крови и тканях.
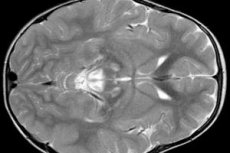
По результатам ЭЭГ выявляют фокальные признаки эпилептической активности. По данным МРТ обнаруживают расширение желудочков мозга, двустороннее поражение мозга, кальцификацию базальных ганглиев (хвостатого ядра, скорлупы, чёрной субстанции, бледного шара). Можно также выявить атрофию больших полушарий и вещества мозга.
При морфологическом исследовании обнаруживают грубые изменения вещества мозга: симметричные очаги некроза, демиелинизации и губчатой дегенерации мозга, преимущественно средних отделов, моста, подкорковых узлов, таламуса, зрительного нерва. Гистологическая картина включает кистозное перерождение мозговой ткани, астроцитарный глиоз, гибель нейронов, увеличение количества митохондрий в клетках. В скелетных мышцах - накопление липидных включений, снижение гистохимической реакции на комплексы 1, 4 дыхательной цепи, субсарколеммальное скопление митохондрий, аномальные митохондрии с дезорганизацией крист. Феномен RRF часто не обнаруживают.
Как обследовать?
Какие анализы необходимы?
Last reviewed: 25.06.2018